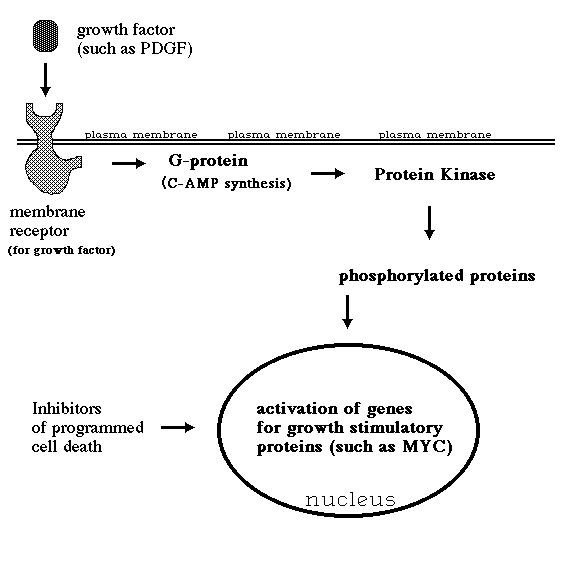
How to cure cancer?
To cure cancer, you need a drug or other treatment that kills cancer cells but doesn't kill normal cells.The difference wouldn't need to have any relation to growth rates, DNA synthesis, or mitotic spindles.
Any difference could be the basis of a cure. Researchers have been very foolish to concentrate efforts so narrowly on growth rates.
At current rates in the USA, 25% of you will get cancer.
Four-fifths of this 25% will die from it.
That's 20 people out of every one hundred.
The cure rate is increasing, but slowly.
____________________________________________________________________________________________
Imagine if the cytoplasm of cancer cells were abnormally acidic, with high concentrations of lactic acid.
Then you could design a drug that selectively activates caspases when pH drops below some threshold level; or that releases a peptide to whose amino acid sequence the patient has been immunized. If any cell contains smallpox peptides, then lymphocytes will stimulate its self-destruction.
Alternatively, if for any reason cancer cells used more glucose than normal cells, some poisonous isotope could be covalently incorporated into glucose, so that the cancer cells would be damaged more than the normal cells, in proportion to the difference in rate of glucose uptake. For example, suppose cancer cells absorbed ten times as much glucose as normal cells, or a hundred times as much.
Next, let's imagine that the acto-myosin cytoskeleton of cancer cells were consistently disorganized, in comparison with normal cells of the same differentiated cell type. What opportunities might that create for selectively tearing holes through their plasma membranes, or causing other physical damage? ____________________________________________________________________________________________
In fact, cancer cells have lots of abnormalities. Fast growth is NOT one of them.
Six actual abnormalities of cancer cells:
-
1) Very acidic cytoplasm (Hydrogen ion and lactic acid concentrations hundreds of time normal).
2) Two hundred times the normal rate of glucose absorption (or more).
3) Disorganization of cytoplasmic actin and myosin.
4) Weakening of traction forces exerted through their plasma membranes. (tenth to hundredth)
5) Ability to crawl from adhesive to (much) less adhesive plastics and other substrata.
6) Inability to rearrange collagen to form organ capsules.
Other abnormalities of cancer cells:
7) Lack of anchorage dependence, the ability of cells to survive and continue dividing without being spread out on a solid substratum. Cancer cells can grow and survive suspended in a gel. Non-cancerous cells will undergo programmed cell death if you culture them on an area of substratum that is smaller that the area they normally occupy when attached to a Petri dish. Nicholas Maroudas and Donald Ingber have done the best research on this.
8) Reduction in contact inhibition of cell locomotion
Directional inhibition of cell crawling where once cell touches another.
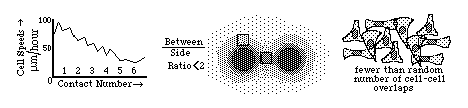
Diagrams of the quantitative experimental tissue culture measurements done by Michael Abercrombie and Joan Heaysman, by which they discovered and proved the existence of contact inhibition of cell locomotion (crawling). By these criteria, they also discovered that cancer cells in tissue culture have less sensitivity to contact inhibition; that seems likely to be at least part of the reason for their increased invasiveness.
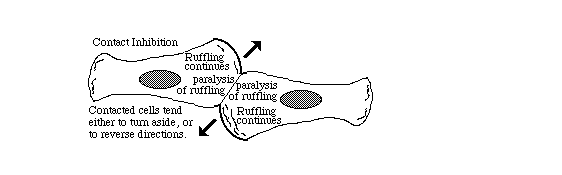
9) Other researchers later used the phrase "contact inhibition" to mean reduced rates of cell growth and mitosis in crowded tissue cultures.Growth and division of cancer cells are less inhibited than equivalent normal cells.
For many years, most scientists confused these two uses of the phrase "contact inhibition" (inhibition of growth as well as locomotion), assumed the same mechanism caused both, and that cancer cells always lacked both (which Abercrombie never claimed).
These mechanisms of inhibition of growth and of locomotion may or may not have the same (or overlapping) mechanisms.
10) Magnetic Resonance Images of cancer cells are dramatically different than normal tissues. This was the subject of the first research paper about MRI. Nobody ever discovered the reason why cancer cells' MRI images are abnormal. Almost no research has been done on the subject.
11) Increased secretion of proteolytic enzymes (metalloproteases, etc.)
12) Loss of differentiated characteristics ("tumor progression")
13) Gain of abnormal combinations of gene transcription
14) Metastasisis transfer of cancer cells from one part of the body to another by breaking free into the lymph or blood (or coelom, or rarely, urine!)
Once cancer cells have begun to metastasize, then it is very much less possible to remove them all surgically.
(Thus, the great importance of early detection)
[Except for lymphoma, etc. which are metastatic very early]
15) Diagnosis of cancer cells by shape and behavior
Cancer cells are usually somewhat abnormal in shape and structure, as seen under the microscope in histological sections. The well known Pap test depends on this fact, although it uses tissue scrapings rather than sections. It is not unusual for cancer operations to begin with the surgeons cutting out a small chunk of the tumor, then sending it down the hall to the histology lab for quick fixation, sectioning, staining and examination by a skilled pathologist. Just by looking at the shapes of the cells and particularly the shapes of the nuclei, the pathologist is supposed to be able to tell whether the tissue is malignant or not.
Books have been written summarizing the criteria used for this purpose. The quotation below is from "The Cytological Diagnosis of Cancer" by Ruth M. Graham, 3rd. edition Saunders, Philadelphia. (Health Sciences Library QZ 241 G 741 1972 ) page 379:
"Whether a cell is malignant or benign is determined by its nucleus; what type of malignant or benign cell it is determined by the cytoplasm."
"In examining a cell, the microscopist should first look at the nucleus and decide whether it is benign or malignant."
"The first feature to look for is the orderly arrangement of the chromatin. Are the chromatin particles of equal size? Are they distributed evenly throughout the entire nucleus? Is the nuclear border smooth and even in thickness? Does each part of the nucleus resemble every other part? If, in the mind's eye, the nucleus were bisected, would the two halves be mirror images of one another? If the answers to these questions are "yes", then one can be sure the nucleus is benign.
On the other hand, if the answers to these questions are "no"; if the chromatin particles differ in size; if they are distributed unequally at the nuclear border, and in the bisected nucleus no part is a mirror image of any other part, then one can be sure that the nucleus is malignant. "
Note that these criteria are entirely empirical (arrived at purely by experience, not based on any theory or other reason for expecting them). Cells with one set of properties always turned out to be malignant in their future behavior (if not removed), while cells with the other sets of properties always turned out to be benign. Mistakes are sometimes made, even mistakes in the direction of diagnosing malignant cells as benign; but given the amounts of money involved in malpractice lawsuits, it seems noteworthy to me that the quotes above could be so general and sweeping. It is almost as if the weather could be accurately predicted from the shapes of clouds, but no one had bothered to find out the physical causation relating the shapes of today's clouds to the occurrence of tomorrow's storms! There has been little or no research into the question of how cell and nuclear shape is related to oncogene function.
The histological organization of cancer cells is also abnormal; the cells' arrangements are "sloppy": they are irregular in shape, sizes and relative positions, as well as slightly out of alignment. What does this imply about the mechanisms that control cell shape, size, relative position, alignment, etc.? Would you prefer to say that the cancerous state interferes with these mechanisms? Or would you say that malignancy results from the disruption or failure of these mechanisms? Your new ideas might really help treatment.