Let's begin with some medical history:
Within living memory, tuberculosis used to be nearly incurable,killed more Americans per year than cancer, and had the same
reputation as cancer for inexorable killing. Tuberculosis was what killed
Anton Chekhov, Robert Louis Stevenson, the mathematician Riemann, all
4 Brontes, Chopin, Emerson, Kafka, Keats, D.H. Lawrence, Thoreau,
Thomas Wolfe, George Orwell and two of President Richard Nixon's brothers,
most around age 40. A diagnosis of tuberculosis used to be an inexorable
death sentence, but this disease then became almost completely curable
with the drugs streptomycin and isoniazid.
I wish I didn't have to admit that resistant strains of tuberculosis have
developed, and have killed millions in poor countries. Nevertheless, the
key points are (first) that breakthroughs do occur, and (second) that medical
breakthroughs are usually based on clever ideas, pursued ruthlessly.
Both for Waksman's discovery of streptomycin in the United States, and
Fleming's discovery of penicillin in Britain, the clever idea was that
micro-organisms might make highly selective poisons that fight germs.
In the search for more antibiotics to fight germs, many chemicals were
discovered that kill cancer cells. None of these have the specificity of
penicillin; they all hurt normal non-cancerous cells but not quite as much
as they hurt cancer cells.
Specificity is the problem: How to find poisons that are specifically poisonous
for germs, or for cancer cells. Specificity always depends on some a
difference between animal cells and bacteria; or a difference between
normal body cells and cancer cells. Any sort of difference can work.
Penicillin gets its specificity from bacterial cell walls; It forms covalent
bonds with the active sites of the enzymes that build bacterial cells walls,
allowing osmotic pressure to burst the bacteria. Animal cells don't have
cell walls, so they aren't harmed.
Streptomycin gets its specificity from differences between procaryotic and
eucaryotic ribosomes. The procaryotic type of ribosomes are distorted more
that the eucaryotic kind.
There are entire books listing selective poisons, and the reasons why they
harm one organism more than others. In every case, the reason is some
difference in the biochemistry, or the physical properties, or their
permeability, or something. An excellent book titled "Selective Toxicity"
lists list hundreds or maybe thousands of specific examples of poisons
that hurt or kill some organisms, but not others. This specificity is always
based on a difference; and based on this book, I think absolutely any
difference can be exploited to create a selective poison.
Fortunately, there are at least a dozen consistent differences between cancer
cells and equivalent normal cells. That implies that two dozen fundamentally
different kinds of anticancer drugs ought to be possible, each based on
exploiting one of the differences, and selectively killing just those cells
that have the difference found in cancer cells.
Unfortunately, almost all the effort has been put into treatments that
interfere with DNA synthesis and mitosis. Not that this effort hasn't
been extremely successfully. Dozens of chemotherapy drugs have been
synthesized or isolated from plants, animals and microorganisms, almost
all of which interfere with cell growth. These have been very successful
in curing the fastest growing kinds of cancer, such as childhood leukemia
and Hodgkin's Disease. Most of the side effects of these drugs also affect
faster-growing normal cells. They produce anemia by harming fast growing
cells in bone marrow; they cause hair loss by harming growing hair cells;
and they produce nausea.
People with slower-growing cancers are less often cured.
Here are some other abnormalities of cancer cells, each of which might be
turned into a new kind of therapy. All you need to do is invent chemicals,
or other treatments, that selectively kill those cells that have one or more
of these abnormal properties.
1) Glucose uptake is much higher than normal in most cancer cells.
Related to this is an abnormally (very) large secretion of lactic acid,
and abnormally low amounts of ATP production by mitochondria. This set of
inter-related abnormalities was discovered around 1930 by Warburg, who had
won the Nobel Prize for previous discoveries, and is one of two (unrelated)
phenomena both called "The Warburg Effect". Excess glucose uptake into cancer
cells is the reason why PET scans can detect cancer, and reliably distinguish
cancer cells from normal cells. I have had several PET scans myself,
and they accurately mapped the tumors. They cost about three thousand
dollars per scan.
The biochemical cause of these phenomena remains unknown, and very little
research has been done on it, because it doesn't fit in with oncogene research.
The best English language textbook on cancer biology doesn't even mention
the Warburg effect. Almost no research has been done about how to kill cancer
cells based on either their anaerobic metabolism, or the excess uptake of
glucose. (Despite very large amounts of research on how to use the phenomenon
to map tumors).
2) MRI images of cancer cells are dramatically different than normal tissues.
This was the subject of the first research paper about MRI.
Nobody ever discovered the reason why cancer cells' MRI images are abnormal.
Almost no research has been done on the subject.
3) The acto-myosin cytoskeleton of cancer cells is disorganized,
and they exert much weaker traction forces as they crawl.
4) Cancer cells can crawl onto less adhesive substrata,
from more adhesive substrata. This is a loss of haptotaxis.
It was discovered by Prof. Barbara Danowski, as part of her PhD research
in this department. She invented the experiment herself. She then did
important research at the University of Pennsylvania, and is now
Professor and Chair of Biology at Union College in New York.
I had never considered the possibility, until she proved it.
5) Defective cell-cycle checkpoint controls.
For example, cancer cells tend to go ahead and copy DNA, even if it is damaged.
Normal cells would delay starting to copy their DNA until
damage to it has been repaired.
6) Excessive inhibition of apoptosis.
In particular, 95% of cases of the "follicular" kind of Non-Hodgkin's lymphoma
are caused by a breakage and incorrect re-joining of the 14th chromosome
and the 18th chromosome, in such a way that the promoter region of the
antibody heavy chain gene is next to a gene named bcl-2. This stands for
b-cell lymphoma two, because it was the second lymphoma-causing gene to be
discovered by looking for chromosome translocations in cancerous lymphocytes
of human patients. Nematodes have a gene almost identical to human bcl-2.
It is needed to inhibit programmed cell death, both in vertebrates and in
nematodes (and probably all animals). Experimenters have even spliced the
human DNA sequence for bcl-2 into nematodes, in place of the nematode's own
gene. The human gene works fine in nematode embryonic development.
When a human b-lymphocyte has this particular chromosome translocation,
it makes bcl-2 protein instead of antibody, and can't undergo programmed
cell death. Although such lymphocytes don't grow or divide any faster than
normal lymphocytes, they do accumulate without limit. Eventually, they
displace your normal lymphocytes and fill up your bone marrow, until you
become anemic, can't make antibodies etc. and die.
Ironically, the treatment is often a chemical designed to kill fast-growing
cells by damaging their DNA. It was assumed the cancerous lymphocytes must
be growing too fast, and that cross-linking their DNA would selectively kill
the fastest growing lymphocytes.(That, itself, isn't very logical.
Why should it kill a cell to prevent it from doing something abnormal?
If they grew slower than normal, would you expect to be able to kill them
by speeding them up?)
Consider the following hypothesis about how chemotherapy really works:
When DNA is damaged by radiation or a drug like cyclophosphamide,
non-cancerous cells detect the damage and slow down their growth,
halting at a check-point until the damaged DNA has been fixed.
The cancerous cells, however, because their check-point controls are broken,
continue to copy DNA and undergo mitosis. This failure to stop is really
what kills them. This is sort of the opposite of what had been assumed.
Cancer cells die because they can't stop, not because they grow too fast.
Any time a drug works, there must be some reason; but the true reason may
be very different than what the drug was designed to do.
7) Lack of anchorage dependence, the ability of cells to survive
and continue dividing without being spread out on a solid substratum.
Cancer cells can grow and survive suspended in a gel. Non-cancerous cells
will undergo programmed cell death if you culture them on an area of substratum
that is smaller that the area they normally occupy when attached to a Petri
dish. Nicholas Maroudas and Donald Ingber have done the best research on this.
8) Increased secretion of proteolytic enzymes(metalloproteases, etc.)
9) Loss of differentiated characteristics ("tumor progression")
10) Gain of abnormal combinations of gene transcription
___________________________________________________________________________________________
There are more kinds in addition; nobody even knows how many more there are,
and almost no research or funding goes toward searching for more.
----------
I) The disease cancer results from a cell of your own body
changing so that it and its mitotic daughter cells
divide and crawl without limit.
It is normal for body cells to grow and divide, and to crawl from place to place.
But cell growth and cell locomotion are normally controlled,
by embryological
mechanisms,which are only partly understood.
"Contact inhibition" means the normal inhibition of cell growth
by crowding, and directional inhibition of cell crawling where
once cell touches another.
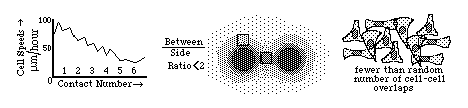
Diagrams of the quantitative experimental tissue culture measurements
one by Michael Abercrombie and Joan Heaysman, by which they discovered
and proved the existence of contact inhibition of cell locomotion (crawling).
By these criteria, they also discovered that cancer cells in
tissue culture have less sensitivity to contact inhibition; that seems likely
to be at least part of the reason for their increased invasiveness.
Other researchers later used the phrase "contact inhibition" to mean
reduced rates of cell growth and mitosis in crowded tissue cultures.
Cancer cells are likewise less inhibited than equivalent normal cells.
For many years, other scientists confused these two uses of the phrase
"contact inhibition" (inhibition of growth as well as locomotion),
assumed the same mechanism caused both, and that cancer cells always lacked
both (which Abercrombie never claimed). Then people over-reacted
in the opposite direction. The inhibition of locomotion now seems
to be caused by prevention of actin fiber assembly near where cells
touch each other. Much more research is needed to find out whether cancer
cellsare more able to continue crawling where they touch other cells,
whether this is related to their uncontrolled growth, or if such
abnormalities can be targeted by new kinds of chemotherapy.
These mechanisms of inhibition of growth and of locomotion
may or may not have the same (or overlapping) mechanisms.
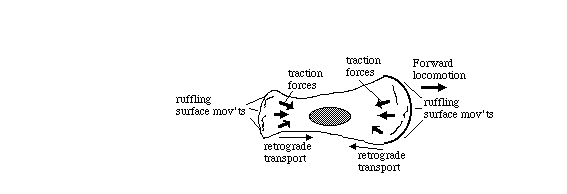
Tissue cells (including body cells in tissue culture) crawl by means
of traction forces exerted behind their leading edges.
Traction
forces are exerted mostly behind areas where the plasma membrane
bends, bubbles and fold irregularly in a process called "ruffling"
(as can be seen in the video time lapse sequences below).
These cell surface movements were was once thought to be peristaltic,
but now are now known to be caused by continual re-assembly of cytoplasmic
actin. Part of contact inhibition is the paralysis of ruffling where
two cells touch each other, as shown in the diagram below.
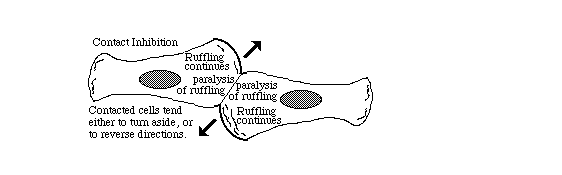
Many tissue cells behave as if actin assembly is somehow turned off where
one cell touches another. Such cells behave as if the mechanism of
locomotion is inhibited next to cell-cell adhesions. Sometimes one or both
cells turns or crawls away from the cell-cell contant; and sometimes new
cell-cell adhesions form, and the adhering parts of both cell margins cease
locomotion and ruffling.
II) The great majority (>95%) of human cancers are caused by
somatic mutations in a few specific genes (called "Oncogenes").
In many other species, large fractions of cancers are caused
by communicable viruses. (Examples include cats and turkeys)
This department gives a very good course specifically about
oncogenes and how they cause cancer.
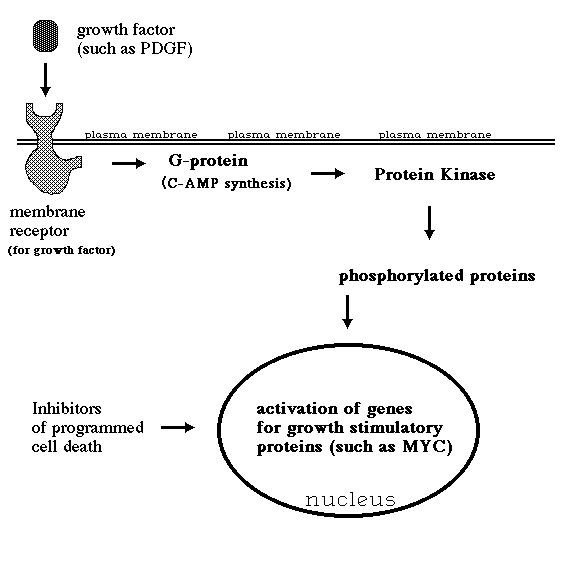
III) Several sexually transmitted papilloma viruses cause
cervical cancer in women. A vaccine has recently been developed
which inhibits infection by these cancer-causing papilloma viruses.
Many newspapers etc. have confused this with 'a vaccine against cancer.'
An editorial in the News and Observer a few years ago "explained"
that the vaccine "Is not yet approved for boys." but didn't ask "Why not?".
These viruses infect both sexes: but the cancers they cause are in females.
What was their reasoning for vaccinating only one sex?
After all, from whom do females catch these viruses?
IV) Cancer cells retain many of the properties of whichever
differentiated cell type they began from:
